the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Apr 2026
| 02 Apr 2026
High-pressure behaviour and elastic constants of 1M and 2M1 polytypes of phlogopite KMg3Si3AlO10(OH)2
Gianfranco Ulian
Francesca Ranellucci
Giovanni Valdrè
In the present work, the elastic properties of both 1M and 2M1 phlogopite polytypes, KMg3Si3AlO10(OH)2 (monoclinic crystal system) were investigated from PV equation of state fitting and by analysis of the fourth-rank elastic tensor. The analysis was performed within the Density Functional Theory framework, using all-electron Gaussian-type orbitals basis sets and the B3LYP functional corrected a posteriori to include long-range interactions (B3LYP-D*). In general, the elastic properties of the two polytypes were strongly anisotropic, with the axial moduli ratio M(a) : M(b) : M(c) being close to . The volume-integrated third-order Birch-Murnaghan equation of state fitting parameters at 0 K were K0=57.9(2) GPa, and V0=489.82(3) Å3 for phlogopite-1M, which were very close to those of the 2M1 polytype, i.e., K0=58.3(1) GPa, ) and V0=978.96(9) Å3. The monoclinic elastic tensors obtained for the two polytypes of phlogopite, which have never been experimentally reported for both minerals so far, were in line with the PV behaviour of the mineral, providing further data related to the directional dependence of the elastic properties and seismic wave propagation. The elastic properties from both PV hydrostatic compression and from the elastic moduli tensor were discussed against the available experimental and theoretical data in the scientific literature, extending the knowledge on this important trioctahedral phyllosilicate.
- Article
(6434 KB) - Full-text XML
-
Supplement
(1118 KB) - BibTeX
- EndNote




Phlogopite [commonly abbreviated with Phl, K(Mg,Fe)3Si3AlO10(OH)2, with Mg Fe ratio greater than 2], is a ubiquitous mica mineral that can be found in different geological environments and pressure-temperature conditions, i.e. igneous, sedimentary, and metamorphic rocks (Icenhower and London, 1995; Trønnes, 2002), and one of the water and potassium reservoirs in the Earth's mantle (Virgo and Popp, 2000; Tutti et al., 2000).
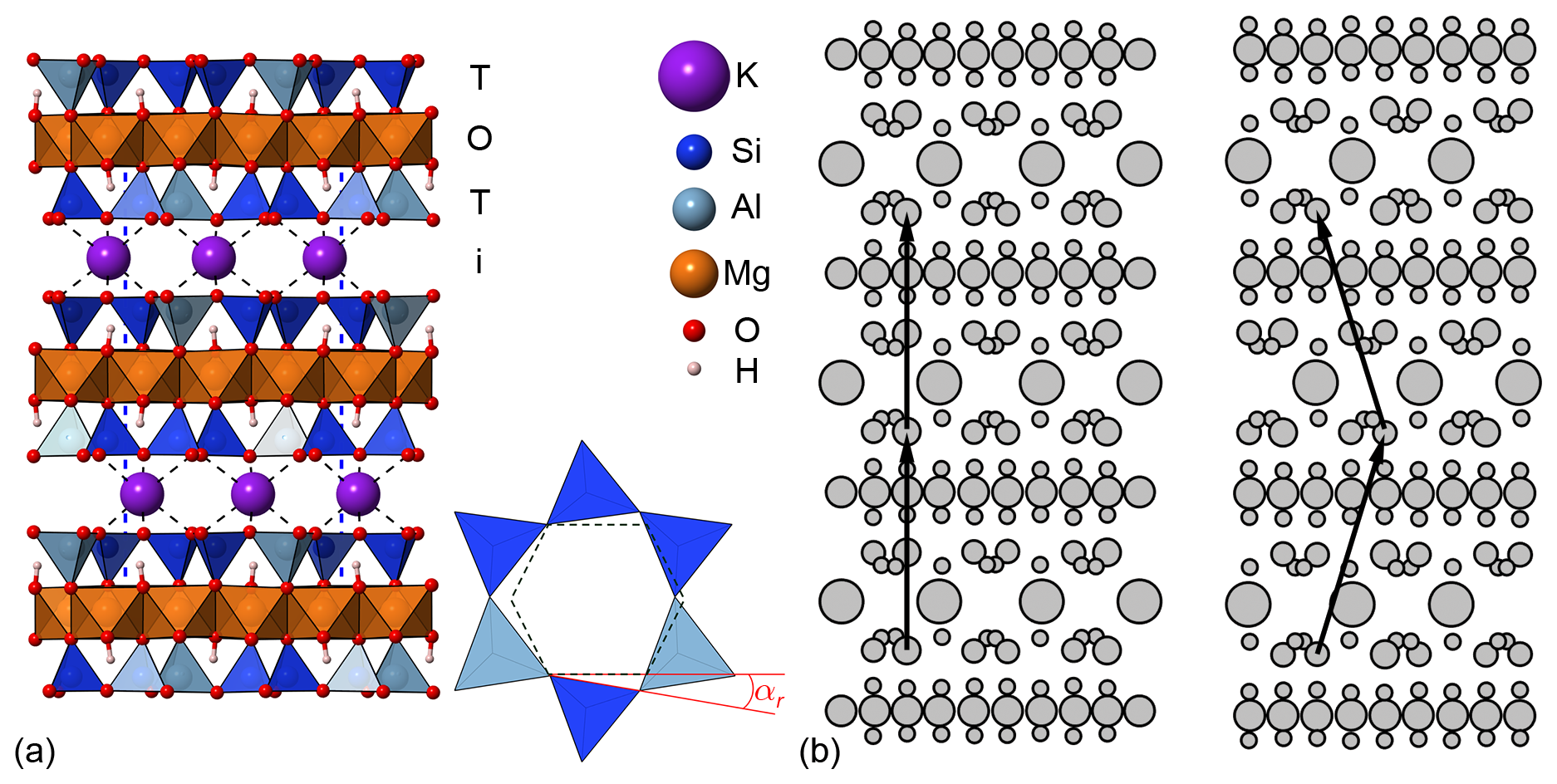
Figure 1(a) Structural model of the phlogopite-2M1 model viewed along the [100] direction. In the bottom right corner, a graphical representation of the tetrahedral rotation angle αr is shown. (b) Schematic view of the stacking of the TOT layers in the 1M (left) and 2M1 (right) polytypes. The black arrows link two points related by symmetry.
From the crystallographic point of view, like other phyllosilicates, phlogopite is made of a 2:1 layer formed by two tetrahedral sheets (labelled as T) that sandwich an octahedral one (O). These T-O-T (or TOT) layers present strong (covalent/ionic) in-plane bonds and a negative charge because of AlIII/SiIV substitutions in the T sheets, which is balanced by potassium cations in the interlayer. Thus, the structure is held together along the c crystallographic axis by a mix of Coulombic (electrostatic) and weak (van der Waals) interactions (Ventruti et al., 2009). Phlogopite presents different polytypes due to different arrangement of the TOT layers along the [001] direction, with the 1M and the 2M1 being the most common in nature both belonging to the monoclinic crystal system (Lacalamita et al., 2012), while a third polytype, 3T (trigonal), is known to be very rare (Gatta et al., 2011). A graphical representation of the most common polytypes is shown in Fig. 1.
Phlogopite is also a widely adopted mineral in the technological field, for instance in the manufacture of ceramics and glasses (King et al., 2000; Ariane et al., 2023) and, more recently, gained attention as a potential dielectric (insulating) material for optical and electronic applications because it presents a band gap of ca. 5–6 eV that can be modulated by microchemical variations, e.g., by FeII/MgII substitutions (Ulian and Valdrè, 2023a).
Micas, and then phlogopite, could be used as geothermobarometers to explain petrogenetic processes occurring in high-pressure and high-temperature (non-ambient) conditions (Guidotti and Sassi, 1976), being also a possible carriers of water and potassium in the Earth's mantle (Sudo and Tatsumi, 1990). As suggested by Melzer and Wunder (2001), since phlogopite may be formed in the mantle wedge that overlies a subducting slab, it could play a relevant role in the element ratios of large ions (K, Rb, Cs) in the subduction zone. Also, the bulk chemical composition of altered rocks such as mica-amphibole-rutile-ilmenite-diopside (MARID) is very similar to the chemistry of phlogopite in an olivine – quartz – alkali ternary [(Mg,Fe)2SiO4–SiO2(quartz)–K2O+Na2O] (alkali) ternary (Sweeney et al., 1993). From the technological perspective, phyllosilicates are considered low-cost insulating materials that could be used in two-dimensional optoelectronic applications, e.g., 2D transistors and heterostructures (Ulian and Valdrè, 2025), thanks to their easy exfoliation, which is related to the perfect cleavage on their (001) planes. Thus, detailed knowledge of the elasticity of phlogopite is of utmost importance for both geological studies, e.g., to explain geophysical observations involving phlogopite-bearing rocks (Geng et al., 2024; Van Reenen et al., 2023; Wang et al., 2023), and to devise new technological uses of this mineral for advanced electronic and optical applications (Cadore et al., 2022). Many experimental studies were focused on the equation of state of phlogopite (Pavese et al., 2003; Comodi et al., 2004; Gatta et al., 2011), but mainly on the 1M polytype. As also observed by Chheda et al. (2014), no measurement of the full monoclinic elastic constant tensor has ever been reported in the scientific literature. The only experimental evidence of this elastic property is related to elastic wave propagation (EWP) experiments that provided just the 5 independent constants, considering the mineral as pseudo-hexagonal (Aleksandrov et al., 1974; Alexandrov and Ryzhova, 1961). The full elastic tensor of phlogopite-1M was only recently provided from ab initio simulations within Density Functional Theory (DFT) at the local density approximation (LDA) and generalized gradient approximation (GGA) levels (Chheda et al., 2014). However, the authors did not employ any correction to include long-range interactions in the physical treatment of phlogopite. Albeit the general consensus is that the TOT layers in micas are held together by the interlayer cations (K+), hence the interaction is mostly of Coulombic (electrostatic) nature, previous works on muscovite and phlogopite showed that the contribution from van der Waals interactions is not negligible in determining the crystal structure, elastic and thermodynamic properties of these minerals (Ulian and Valdrè, 2015a; Ulian and Valdrè, 2023a). Furthermore, no studies on the elastic properties of phlogopite-2M1 were ever carried out and reported. The differences in the elastic behaviour of the two phlogopite polytypes could be indeed subtle, especially in terms of the second-order elastic moduli tensor, where small variations in the directional properties between the 1M and 2M1 are hypothesised.
For all these reasons, the present work is focused on the elastic properties of phlogopite, considering both 1M and 2M1 polytypes. To this aim, ab initio DFT simulations were performed (i) to understand the compressional behaviour of the mineral through an equation of state fitting, (ii) to obtain the full monoclinic elastic moduli and (iii) to assess the role of polytypism on the elastic behaviour of phlogopite. All the data were compared and discussed with both experimental and theoretical results found in the scientific literature to extend the knowledge on this important phyllosilicate.
As previously introduced, the simulations reported in the present work were carried out within the Density Functional Theory framework. In detail, the hybrid B3LYP functional (Becke, 1993; Lee et al., 1988), which includes 20 % of exact Hartree-Fock exchange and some non-local contribution to the exchange-correlation terms, was employed throughout the present study. B3LYP is considered a suitable choice when dealing with the simulation of the structural and elastic properties of minerals due to its high accuracy, and it was already adopted with success for the description of several other minerals (Prencipe et al., 2009; Ottonello et al., 2010; Belmonte, 2017; Ulian and Valdrè, 2023b, 2019) and phlogopite (Ulian and Valdrè, 2023a). However, since most generalized-gradient approximation functionals, including their hybrid counterparts, do not adequately include van der Waals (long-range) interactions, the DFT-D2 correction (Grimme, 2006) was adopted, employing the parameters of Civalleri and co-workers (2008) for the B3LYP functional (B3LYP-D* method). According to the proposed methodology, the original B3LYP-D2 parameters were modified as s6=1, the van der Waals radius of the hydrogen atom, Rvdw(H), was set to 1.30, and the RvdW of the heavier atoms were scaled by a factor of 1.05, while the C6 values were the same proposed in the DFT-D2 scheme (see Table S1 in the Supplement). This approach was used in previous studies on the high-pressure behaviour of talc (Ulian et al., 2014, Ulian and Valdrè, 2015c), pyrophillite (Ulian and Valdrè, 2015b), muscovite (Ulian and Valdrè, 2015a), resulting in data in very good agreement with experimental data. The all-electron Gaussian-type orbitals (GTO) used to construct the Kohn-Sham orbitals within the linear combination of atomic orbitals (LCAO) approach were the same used in a recent work on the 1M polytype of phlogopite (Ulian and Valdrè, 2023a). Si, Al, Mg, K, O and H atoms were described with 88-31G* (Nada et al., 1996), 85-11G* (Catti et al., 1994), 8-511d1G (Valenzano et al., 2007), 86-511G (Dovesi et al., 1991), 8-411d11G (Valenzano et al., 2006), and 3-1p1G (Gatti et al., 1994), respectively.
The accuracy of the Coulomb and exchange series was controlled by five thresholds set to 10−8 (ITOL1 to ITOL4) and 10−16 (ITOL5) for the structural relaxation procedures, whereas the convergence on the total energy was set to 10−8 Hartree. The reciprocal space was sampled with a Monkhorst–Pack mesh (Monkhorst and Pack, 1976), corresponding to 39 independent k points.
The lattice and internal geometry were optimised within the same run using a numerical gradient for the unit cell parameters and an analytical gradient for the atomic positions. The Hessian matrix was upgraded with the BFGS algorithm (Broyden, 1970b, a; Fletcher, 1970; Goldfarb, 1970; Shanno, 1970). The tolerances for the maximum allowed gradient and the maximum atomic displacement were set to 10−5 Ha bohr−1 and bohr, respectively.
All the simulations were performed with the CRYSTAL17 code (Dovesi et al., 2018), whereas the QUANTAS software (Ulian and Valdrè, 2022, 2024a) was employed to post-process the elastic data. Graphical representations have been carried out with the molecular graphics program VESTA (Momma and Izumi, 2008).
The initial model of the crystal structure of 2M1- phlogopite was created from the single-crystal X-ray diffraction data of Lacalamita et al. (2012), which was subsequently optimised within the B3LYP-D* level of theory. The mineral belongs to the monoclinic system (space group C2/c), presenting two independent tetrahedral sites (labelled as T) and two octahedral sites (M), with partial occupancies of the cationic sites with several possible atoms. For example, the TO4 tetrahedral sites in the unit cell are generally occupied by about 70 % by Si and 30 % by Al (Laurora et al., 2007; Lacalamita et al., 2012), which can be translated in sites being randomly occupied by aluminium. However, as also explained in a previous study on the 1M polytype (Ulian and Valdrè, 2023a), theoretical modelling requires each site to be deterministically occupied by a single kind of atom; in the example cited above, a T site could present either Al or Si but not both at the same time. Thus, to preserve the monoclinic lattice of phlogopite when the AlIII/SiIV substitutions are included in the structure, the 2M1 polytype with formula KMg3(AlSi3)O10(OH)2 was described with the P2/c space group (23 inequivalent atoms, 88 atoms in the unit cell, Z=4 unit formulas), which is a sub-group of the C2/c symmetry. Within the P2/c space group, phlogopite-2M1 presented four inequivalent T sites (SiIV in T1–T3, AlIII in T4) and three non-equivalent O sites (M1–M3). Hence, each T site was related to four SiO4 or AlO4 tetrahedra, whereas all octahedral sites are related to two MgO6 octahedra.
3.1 2M1-Phlogopite crystal structure
Table 1 reports the lattice parameters, tetrahedral (Tthick) and octahedral (Mthick) sheet thicknesses, interlayer distance (Ithick), and selected polyhedral properties of the 2M1 phlogopite polytype, obtained from DFT simulations with (B3LYP-D*) and without (B3LYP) the correction to include the effects of long-range interactions. Previous XRD refinements are also shown in Table 1 for a direct comparison.
Table 1Equilibrium geometry of 2M1-phlogopite as obtained from DFT/B3LYP and B3LYP-D*, compared to previous experimental refinements.
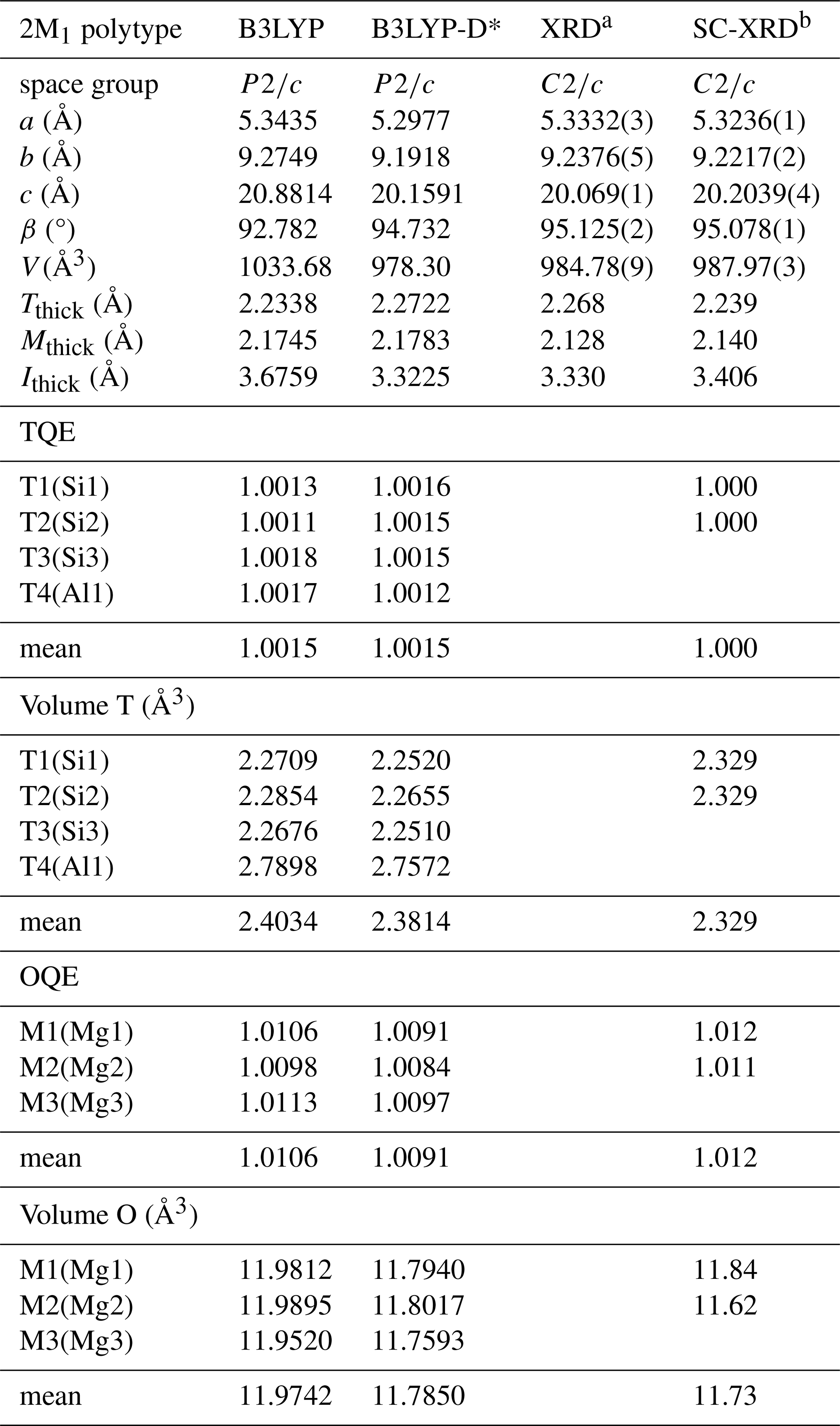
a X-ray diffraction data of Laurora et al. (2007). b single-crystal XRD refinement of Lacalamita et al. (2012) performed on sample BU1_14. Tthick, Mthick, and Ithick are tetrahedral sheet thickness (calculated from the z coordinates of the basal and apical oxygen atoms), the octahedral sheet thickness (calculated from the z coordinates of the apical and hydroxyl O atoms) and interlayer thickness (calculated from the z coordinates of the basal oxygen atoms), respectively. TQE and OQE are the tetrahedral and octahedral quadratic elongations, respectively (Robinson et al., 1971). T and M represent the generic tetrahedral and octahedral sites, respectively.
The simulations at the DFT/B3LYP-D* level of theory are in good agreement with the experimental crystallographic data reported in literature, showing a small underestimation of the unit cell volume (ΔV ), which is the result of a slight expansion of the tetrahedral and octahedral sheet thicknesses (ΔTthick=1.5 % and ΔOthick =1.8 %, respectively) and a contraction of the interlayer distance (ΔI). These results for the 2M1-phlogopite are in agreement with previous simulations of the 1M polytype (Ulian and Valdrè, 2023a) and other phyllosilicates and layered minerals (Ulian et al., 2013; Ulian and Valdrè, 2015a, b, 2019), where the inclusion of long-range interactions in the physical treatment is of utmost importance to properly simulate the crystal-chemistry and properties of this kind of structure. When the DFT-D2 correction is not implemented in the simulation framework, a dramatic increase in the unit cell volume can be noted (+6 % with respect to the DFT/B3LYP-D* results, +5 % from the XRD data), which is the result of the increase of the lattice parameters a (+0.9 %), b (+0.9 %), and c (+3.6 %) and the shrinking of the β angle (−2.1 %). Considering the absence of any thermal effects in the Density Functional Theory simulations, the overestimation of the lattice constants and unit cell volume is a further sign of the relevant role played by van der Waals interactions in micas.
3.2 Compressional behaviour of phlogopite 1M and 2M1 polytypes
The compressional behaviour of phlogopite was modelled considering 10 different unit-cell volumes, both smaller (compressed, 7 volumes) and larger (expanded, 3 volumes), within 88 % and 108 % of the equilibrium geometry volume (Veq). Then, the internal coordinates and lattice parameters were optimized keeping the selected volume fixed during the procedure, constraining the space group of 1M-phlogopite (P2) and 2M1-phlogopite (P2/c), a procedure known as “symmetry preserving, variable cell-shape structure relaxation” that was proposed by Pfrommer et al. (1997). The equilibrium geometry of 1M-phlogopite was retrieved from the work of Ulian and Valdrè (2023a) that was obtained with the same computational settings here adopted for the 2M1 polytype.
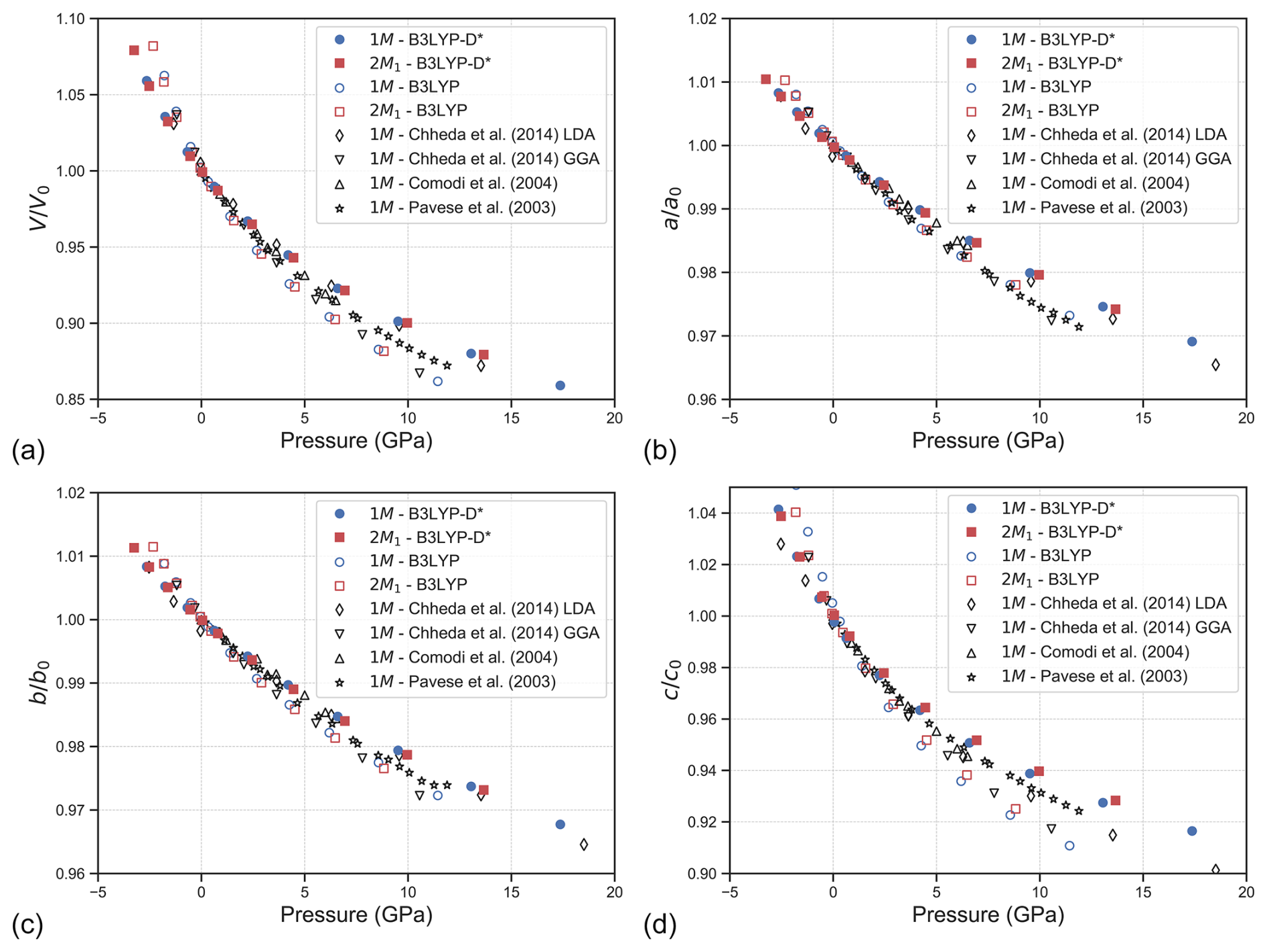
Figure 2Evolution of normalised (a) unit cell volume , (b) , (c), and (d) as a function of pressure for the phlogopite polytypes 1M and 2M1. The experimental data of Pavese et al. (2003) and Comodi et al. (2004) are reported for a comparison.
The dependence of the total energy E of the mineral at the different volumes V, i.e., the E(V) curve, was described in terms of a volume-integrated 3rd-order Birch-Murnaghan equation of state, BM3 (Birch, 1947), as reported by Hebbache and Zemzemi (2004):
where η is a dimensionless parameter, K0 is the bulk modulus at 0 K, K′ its pressure first derivative and V0 the volume at zero pressure. The pressure values at each unit cell volume (reported in Tables S2 and S3 for the 1M and 2M1 polytypes, respectively) were calculated using the fitting parameters K0, K′ and V0 obtained from the previous equation and employed in the well-known P-V formulation of the BM3 (Birch, 1947):
with P0 the reference pressure (0 GPa). In Table 2, the BM3 equation of state parameters for both 1M and 2M1 phlogopite are reported. A graphical representation of the normalised volume and axis lengths as a function of calculated pressure is presented in Fig. 2., alongside the experimental results from X-ray diffraction (Comodi et al., 2004; Pavese et al., 2003) and the theoretical calculations of Chheda et al. (2014) carried out at the LDA and GGA level of theory.
A direct comparison between the different experimental and computational approaches is however possible on the phlogopite-1M, which is better depicted in Fig. S1 (Supplement) considering the absolute values of the cell volume and lattice vectors. In general, compared to the high-pressure XRD refinements, the crystal structure from the DFT/B3LYP-D* approach well agrees with the results of Pavese et al. (2003) at low pressure (P<5 GPa), however there is a slight overestimation of the unit cell volume at high pressure (see Figs. 2a and S1a). Analysing the lattice vectors, it can be noted that the a and b lengths are very close to the experimental ones at each pressure state, whereas the c-axis shows a stiffer behaviour and an overestimation that increases with pressure. By considering the results of Comodi et al. (2004), the present B3LYP-D* data are slightly underestimated in terms of unit cell volume, which is due to the lower values of the a- and b-axes, whereas the evolution of the clattice parameters is correctly described by the proposed computational approach (see Fig. S2b–d). The stiffer behaviour of both polytypes at higher pressures elucidated by the trend is related to the static conditions of the simulations, i.e., the zero-point and thermal effects were neglected. It is suggested that the use of the quasi-harmonic approximation, which is however beyond the scope of the present work, could reduce the mismatch between the theoretical and experimental data.
It is interesting to note that if the long-range interactions are not included in the theoretical framework, the B3LYP functional leads to a general overestimation of the unit cell volume and lattice parameters. The a and b vectors are closer to the results of Comodi and collaborators (2004) than those of Pavese et al. (2003), however, the c lattice parameter is severely overestimated when compared to both XRD results. All these considerations are reflected in the PV fitting using the BM3 equation of state, where the B3LYP-D* approach resulted in a small overestimation of the bulk modulus K0 when compared to the experimental data (see Table 2), and the theoretical K' and V0 values between the ranges obtained from the XRD refinements. Conversely, the B3LYP data shows a large underestimation of the bulk modulus of about 18 GPa, and an overestimation of the cell volume.
Table 2Equation of state parameters and associated standard deviations (σ) obtained for the 1M and 2M1 polytypes of phlogopite.
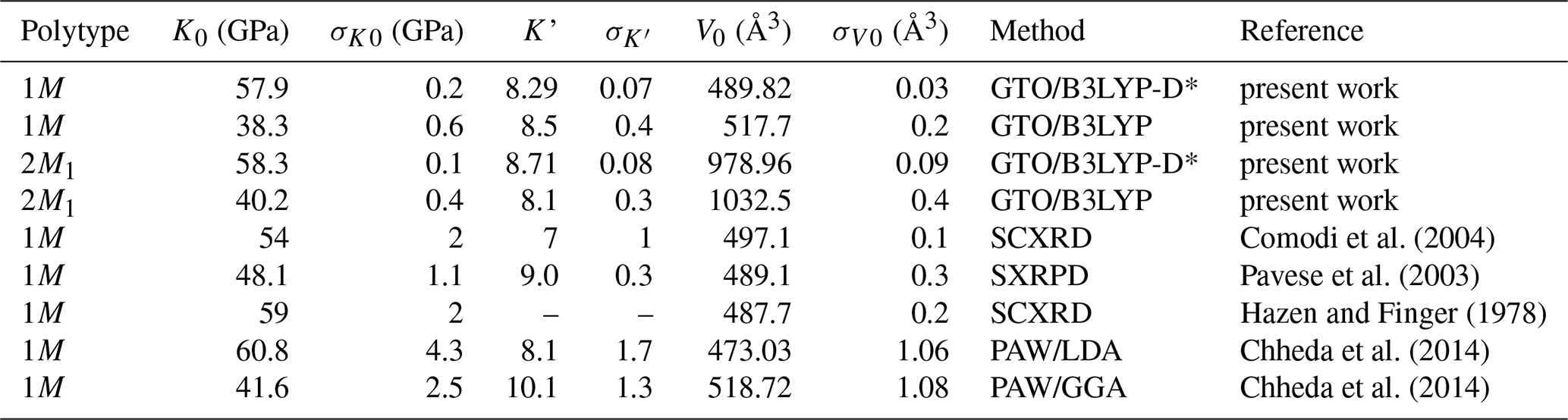
Notes: GTO – Gaussian-type orbitals; PAW – projector-augmented wave; SCXRD – single-crystal X-ray diffraction; SXRPD – synchrotron X-ray powder diffraction.
Compared to previous theoretical results, the crystal structure of the phlogopite 1M polytype and the related PV fitting obtained from the uncorrected B3LYP functional are almost identical to the GGA values of Chheda et al. (2014). Instead, the B3LYP-D* approach, which includes the modified DFT-D2 correction, provided crystallographic results that are between those calculated using the LDA (lower bound in Fig. S1) and the GGA functionals (upper bound). Interestingly, the LDA approach led to a correct description of the c lattice parameter with pressure, whereas the generalised-gradient approximation functional resulted in the same overestimation noted with the B3LYP approach. Thus, GGA and hybrid B3LYP functionals provide an adequate description, i.e., within the experimental information, of the a and b lattice parameters, where strong ionic/covalent bonds are responsible of the TOT structure; though, both approaches fail in properly describing the c-axis behaviour. This is a further confirmation of the hypothesis that the forces acting along the [001] direction of micas are not only electrostatic/ionic, but there is also a contribution from weak van der Waals interactions (Ulian and Valdrè, 2015c, 2023a), as demonstrated using the DFT-D2 correction. The authors are aware that the B3LYP-D* approach, whose DFT-D2 parameters were empirically recalibrated on molecular crystals (Civalleri et al., 2008), is just one of the possible methods to properly treat long-range interactions, and other more ab initio methods could be employed, e.g., the vdW functionals developed of Dion et al. (2004). However, the proposed theoretical framework was already suitable to provide new insights into this fundamental topic regarding the bonding nature of mica phyllosilicates.
The axial compressibility, M0, was obtained using a linearized form of the third-order Birch-Murnaghan equation of state, as described by Angel et al. (2014), which was also employed to re-calculate the experimental compressibility reported by Pavese et al. (2003) and Comodi et al. (2004) including the uncertainties on pressure and lattice parameters. The results are shown in Table 3, alongside the theoretical results of Chheda and collaborators (2014). The results are consistent for all the simulations, as the bulk modulus calculated from the axial compressibility is within 2.64 % (1M) and 1.67 % (2M1) from the K0 value obtained from the equation of state fit. In general, it can be noted that the K0 values are almost the same between the two polytypes. However, the 1M and 2M1 phlogopite models showed a slightly different behaviour in terms of axial compression. In fact, while the M0(a) and M0(b) values are the same between the two phlogopite models, the M0(c) modulus is significantly higher in the 2M1 polytype, with a stiffness increase of about 10 % with respect to the 1M one. Similar results were obtained by Chheda et al. (2014) from DFT simulations and by Pavese et al. (2003) from XRD experiments. Also, the recalculated values of Comodi et al. (2004) with the linearized formulation lead to a bulk modulus within 0.57 % the value obtained from the PV EoS fitting.
Table 3Axial compressibility parameters obtained from linearized equation of state fit.
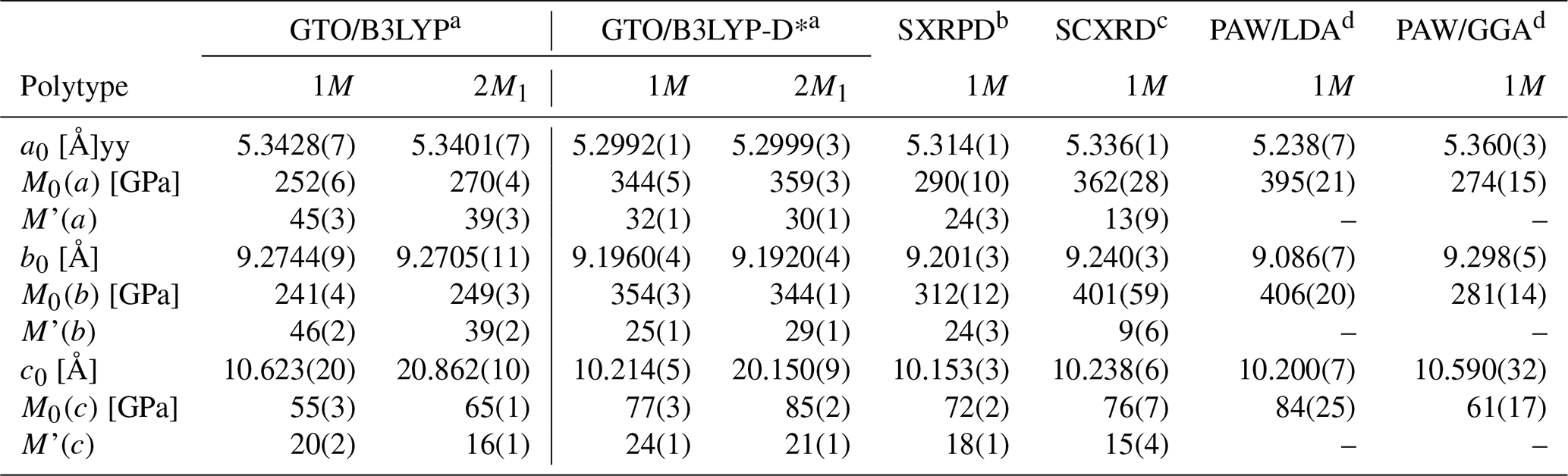
a Present work. b Pavese et al. (2003). c Comodi et al. (2004). d Chheda et al. (2014). GTO – Gaussian-type orbitals; PAW – projector-augmented wave; SCXRD – single-crystal X-ray diffraction; SXRPD – synchrotron X-ray powder diffraction.
Crystal structure data of both 1M and 2M1 polytypes are reported in Tables S2 and S3, respectively, providing further insights into the pressure effects on the phlogopite internal geometry.
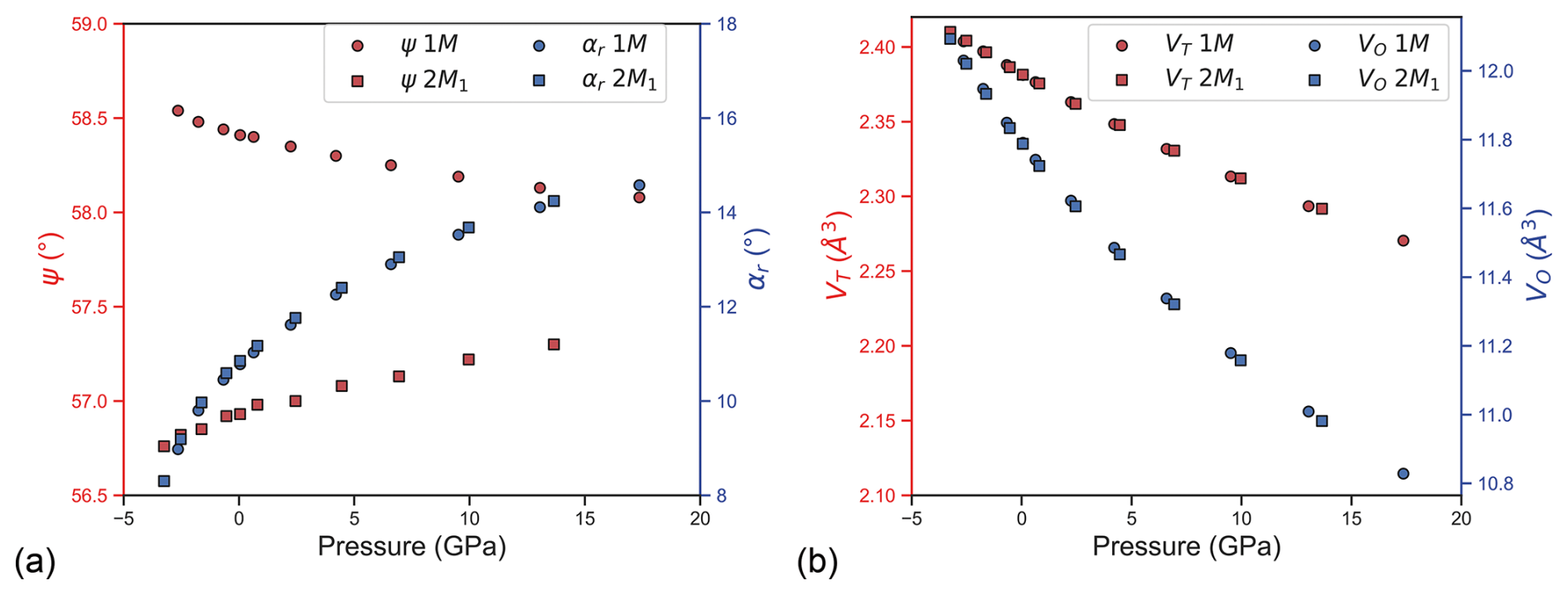
Figure 3(a) Variation as a function of pressure of the octahedral flattening angle ψ (red symbols) and tetrahedral rotation angle αr (blue symbols) in the 1M and 2M1 phlogopite models. (b) Evolution of the tetrahedral TO4 and octahedral MO6 polyhedral volume with pressure.
The TO4 tetrahedra showed a stiff behaviour, with zero-pressure bulk moduli equal to 282(3) and 285(2) GPa for the 1M and 2M1 polytypes, respectively, whereas the octahedral MO6 units were more compressible, presenting a bulk modulus of 143(1) GPa for both phlogopite polytypes (Fig. 3a). This means that the TOT layer of the two minerals presented similar elastic properties. As also explained in previous works on phyllosilicates (Comodi et al., 2004; Chheda et al., 2014; Ulian and Valdrè, 2015c), the variation of the tetrahedral rotation angle αr (Donnay et al., 1964b, a) is the main mechanism that accommodated the compression of the mineral. The αr value is greater than 0 for both phlogopite polytypes at equilibrium, which means that the TO4 mesh is not hexagonal (ideal) but forms a di-trigonal ring (see Fig. 1), and αr increases with pressure as reported in Fig. 3b. This trend is in line with the simulations performed by Chheda and collaborators (2014), and with the general compressional behaviour of trioctahedral micas reported by several authors (Comodi and Zanazzi, 1995; Gatta et al., 2015, 2011; Pavese et al., 1999, 2003; Pawley et al., 2002). In addition, it is interesting to note that the octahedral flattening angle ψ became smaller by increasing pressure in the 1M polytype, whereas a direct proportionality was observed for the 2M1 one.
3.3 Elastic constants and derived quantities
To provide a complete analysis of the elastic behaviour of phlogopite polytypes, the second-order elastic tensor of the two models was also calculated. The approach relies on stress-strain relationships based on total energy E(V, ε) calculations through a Taylor expansion in terms of the strain components truncated at the second order:
where ε is the strain, σ is the stress, C is the 6×6 matrix representation of the elastic tensor (Voigt's notation), V is the volume of the strained unit cell and V0 is the volume at equilibrium. In this expression, α, β=1, 2, 3, … 6. The adiabatic second-order elastic moduli are related to the second derivatives of the total energy E on the strain according to:
The Cαβ values were calculated by imposing a certain amount of strain ε along the crystallographic direction corresponding to the component of the dynamical matrix. This procedure is automated in the CRYSTAL code (Perger et al., 2009), and in the present simulations the default values that control the calculation of the elastic moduli were employed. To calculate the elastic tensor, the crystal was oriented with the b and c crystallographic axes parallel to the y and z Cartesian axes, respectively. Within this crystal orientation and considering the monoclinic lattice, the 6×6 matrix C of the elastic moduli in Voigt's notation is given by:
whose values are reported in Table 4, considering both B3LYP and B3LYP-D* approaches. As expected, the inclusion of long-range interactions leads to a stiffer behaviour of phlogopite, especially for the diagonal terms Cαα.
Table 4Elastic moduli Cαβ (in GPa) of phlogopite polytypes obtained in the present DFT simulations, compared to previous theoretical and experimental data.
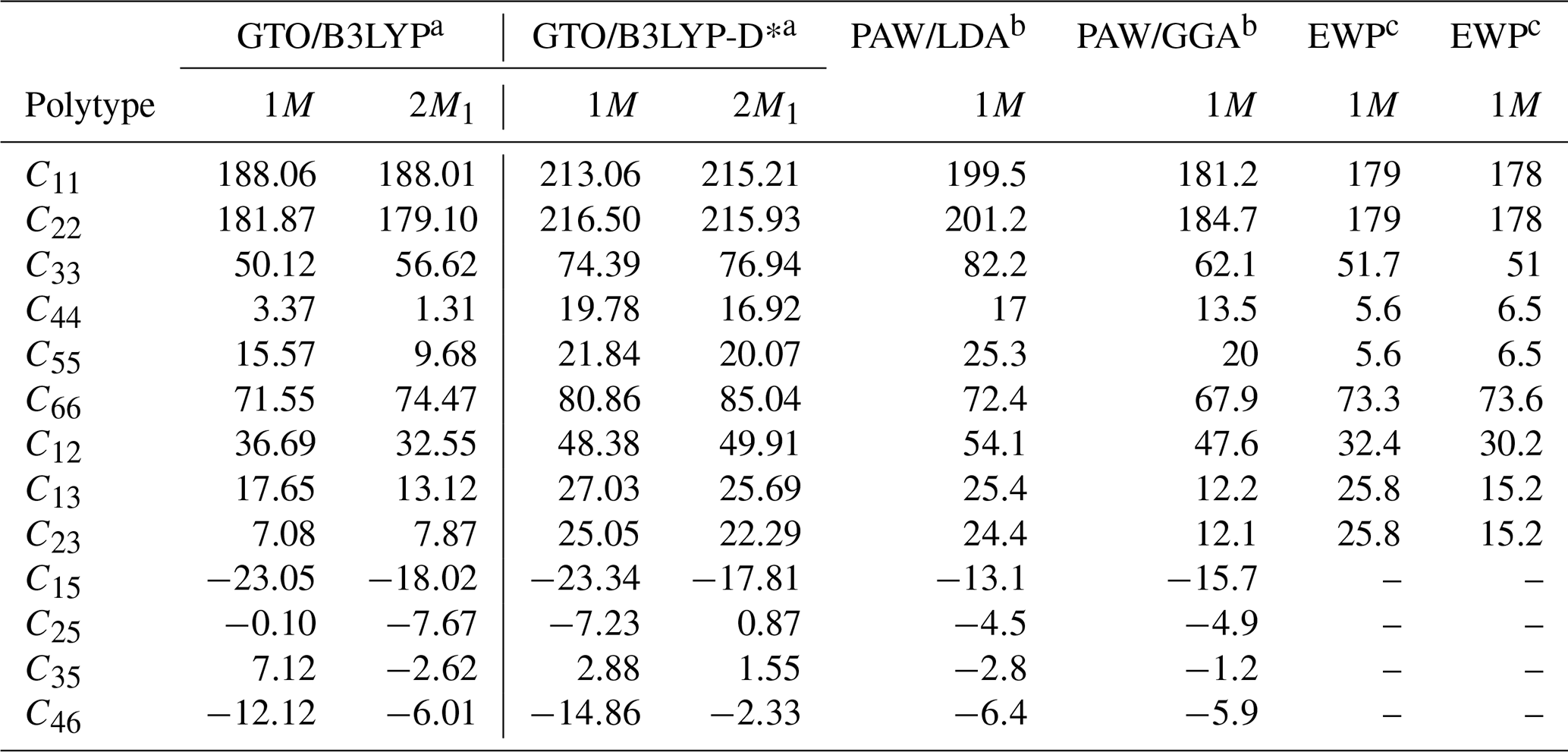
Notes: GTO – Gaussian-type orbitals; PAW – projector-augmented wave; EWP – Elastic wave propagation. a Present work. b Chheda et al. (2014). c Aleksandrov et al. (1974).
Very few experimental data were reported in the scientific literature because suitable single crystals of phlogopite are rare, and many of these specimens are required for a complete characterisation of the elastic tensor of low-symmetry phases. Nevertheless, the present simulations are in good agreement with the experimental results of Alexandrov and Ryzhova (1961) and Aleksandrov et al. (1974), where the authors measured the elastic moduli from two crystals, one from the Slyudyanka and one from the Aldan region, near Lake Baikal (Russia). It is worth noting that only 5 independent elastic tensor components related to the hexagonal symmetry were reported instead of the 13 moduli required for the actual monoclinic symmetry of phlogopite. Also, two pairs of elastic moduli were equal, i.e., C11= C22 and C44=C55, and the off-diagonal components C15, C25, C35 and C46 were zero due to symmetry constraints. The present theoretical results at the B3LYP-D* level of theory indeed showed that C11≈C22 and C44≈C55, suggesting the near-isotropic elastic behaviour of the basal plane. Vaughan and Guggenheim (1986) measured the the full monoclinic elastic tensor for the dioctahedral equivalent of phlogopite, i.e., muscovite KAl2Si3AlO10(OH)2, finding a similar elastic behaviour with a small, but not insignificant, deviation from the hexagonal symmetry. Within this computational framework, the calculated Cαα moduli of phlogopite are overestimated by about 31 GPa (α=1, 2, 3) and 13 GPa (α=4, 5, 6) on average, but the trends of the diagonal components and are the same as those experimentally obtained from elastic wave propagation experiments (Aleksandrov et al., 1974; Alexandrov and Ryzhova, 1961). The uncorrected B3LYP functional resulted smaller absolute deviations from the experimental data (ca. 4 and 2 GPa for the uniaxial and biaxial tensor components, respectively) and the same relationship; however, the C44 modulus was smaller than the C55 one, and also much smaller than the one calculated at the B3LYP-D* level. Regarding the off-diagonal components, both approaches resulted in C12>C13, although the uncorrected functional showed C13>C23, while the inclusion of the van der Waals interactions provided C13≈C23, which is in better agreement with the experimental evidence.
The different performance of the B3LYP and B3LYP-D* approaches in calculating the diagonal and off-diagonal components of the stiffness tensor depends on the underlying physics of the elastic response. The diagonal elastic constants are dominated by the response of the strongly bonded T–O–T layers. When dispersion is neglected (B3LYP), long-range interactions are underestimated, leading to an overexpanded interlayer region. In Gaussian basis-set calculations, this partly compensates the overestimation of forces caused by Pulay stress (as discussed below), producing an accidental error cancellation and a seemingly better agreement for the diagonal terms. In contrast, the off-diagonal terms describe shear-normal coupling and interlayer-intralayer stress transfer, which are very sensitive to the correct description of weak interactions. Without dispersion (B3LYP), the layers are too decoupled, and the coupling terms Cαβ (α≠β) are underestimated or show incorrect relative magnitudes. Including dispersion (B3LYP-D*) restores realistic interlayer distances and mechanical coupling, improving the prediction of the off-diagonal elastic constants. This further confirms that the proper treatment of long-range interactions is relevant for the fundamental analysis of the elastic properties of layered silicates, especially when using Gaussian-type orbitals basis sets.
The present results at the B3LYP(-D*) level of theory are also in line with the simulations performed by Chheda and collaborators (2014) using LDA and GGA functionals and plane-waves basis sets, albeit some notable differences are present (see Table 4). Considering the cited work, it is known that the local density approximation generally underestimates lattice vectors and overestimates the forces, resulting in a stiffer behaviour of simulated phases. Conversely, GGA functionals are generally underbinding, hence the calculated elastic properties can be slightly underestimated. Then, hybrid functionals like B3LYP, which include a small fraction of the exact Hartree-Fock exchange to the total energy of the system, are expected to provide results that are between the LDA/GGA extremes if, and only if, other computational parameters (basis set type and quality, k-point sampling, converge criteria, etc.) are the same. Another source of discrepancy between the present elastic results and those of Chheda et al. (2014) is indeed the basis set type. Localised Gaussian-type orbitals generally provide slightly larger Cαβ values than those obtained with plane-waves basis sets in density functional theory simulation because of the Pulay forces that arise from the Hellmann-Feynman theorem and the use of atom-centred GTOs. As previously explained (Ulian et al., 2021; Ulian and Valdrè, 2018), the Pulay forces are associated with the basis set superposition error (BSSE), which is due to the incompleteness of GTOs and that artificially increases the calculated stress/force within a solid phase. The authors are aware that an approach like the geometrical counterpoise (gCP) method (Brandenburg et al., 2013; Kruse and Grimme, 2012) devised for molecule and molecular crystals could at least partially remove this BSSE-related error. However, the current implementation is limited to some general-purpose GTO basis sets that do not include the one employed in the present work, and a thorough analysis of its suitability for inorganic crystals and minerals is still ongoing (Ulian and Valdrè, 2024b). Besides the Pulay stress, the general overestimation of the theoretical results, both the present one and those of Chheda et al. (2014), could be also due to the athermal conditions, i.e., the DFT simulations were carried out at 0 K without zero-point energy and any thermal contribution, while the elastic wave propagation measurements are typically carried out at room temperature (about 300 K). Regarding the present work, the use of gCP and including temperature effect, e.g., with the quasi-harmonic approximation, can provide results that are in more agreement with the experimental findings.
For practical applications, it is commonly preferred the use of the polycrystalline averages, namely the bulk (K) and shear (μ) moduli, calculated with the Voigt–Reuss–Hill approach (Hill, 1952; Nye, 1957) according to the formulas:
where are the component of the compliance tensor, i.e., the inverse of elastic moduli tensor C, and the subscripts V and R indicate the Voigt (upper) and Reuss (lower) bound of the isotropic elastic properties. The Young's modulus E and the Poisson's ratio υ were calculated from the following relations:
and
The polycrystalline mechanical properties were reported in Table 5. The KR values obtained for both phlogopite 1M and 2M1 polytypes were 58.8 and 59.4 GPa, respectively, which are consistent with those calculated through the BM3 fitting procedure (57.9 and 58.3 GPa, respectively).
The elastic anisotropy of phlogopite was described using the universal anisotropic index, AU, as proposed by Ranganathan and Ostoja-Starzewski (2008):
For all but isotropic systems, for which AU=0, the ratio between the Voigt and Reuss moduli is not equal to unity, thus AU>0. The percentage anisotropy in compression (AK) and shear (Aμ) was also calculated according to the following formulas (Ranganathan and Ostoja-Starzewski, 2008):
The anisotropies obtained via these expressions were reported in Table 5, showing that the AU values of the two phlogopite polytypes are very similar (about 3.8 at the B3LYP-D* case), and between the values calculated from the theoretical results of Chheda and collaborators (2014) using the LDA (2.9) and GGA (4.4) functionals. There is a slight discrepancy between the different DFT approaches and the experimental results (AU=11.8), which is related to the high anisotropy of the shear moduli. It is suggested that the high ratio is probably due to the very small values of the C44=C55 elastic tensor components that were experimentally measured by Aleksandrov et al. (1974) with elastic wave propagation methods. Similar figures were found for the bulk and shear anisotropies, with AK≈14 % and Aμ≈26 % calculated at the B3LYP-D* level of theory, intermediate values between those obtained using LDA and GGA functionals by Chheda and co-workers (2014). Conversely, the uncorrected B3LYP functional led to an anomalously high anisotropy index (35.2) due to the very large difference between the Voigt and Reuss shear moduli (Aμ≈77 %).
Table 5Isotropic elastic properties (bulk K, shear μ, and Young's E moduli, and Poisson's ratio υ) of phlogopite, alongside the universal anisotropy index AU and the percentage of anisotropy of the bulk (AK) and shear (Aμ) moduli.
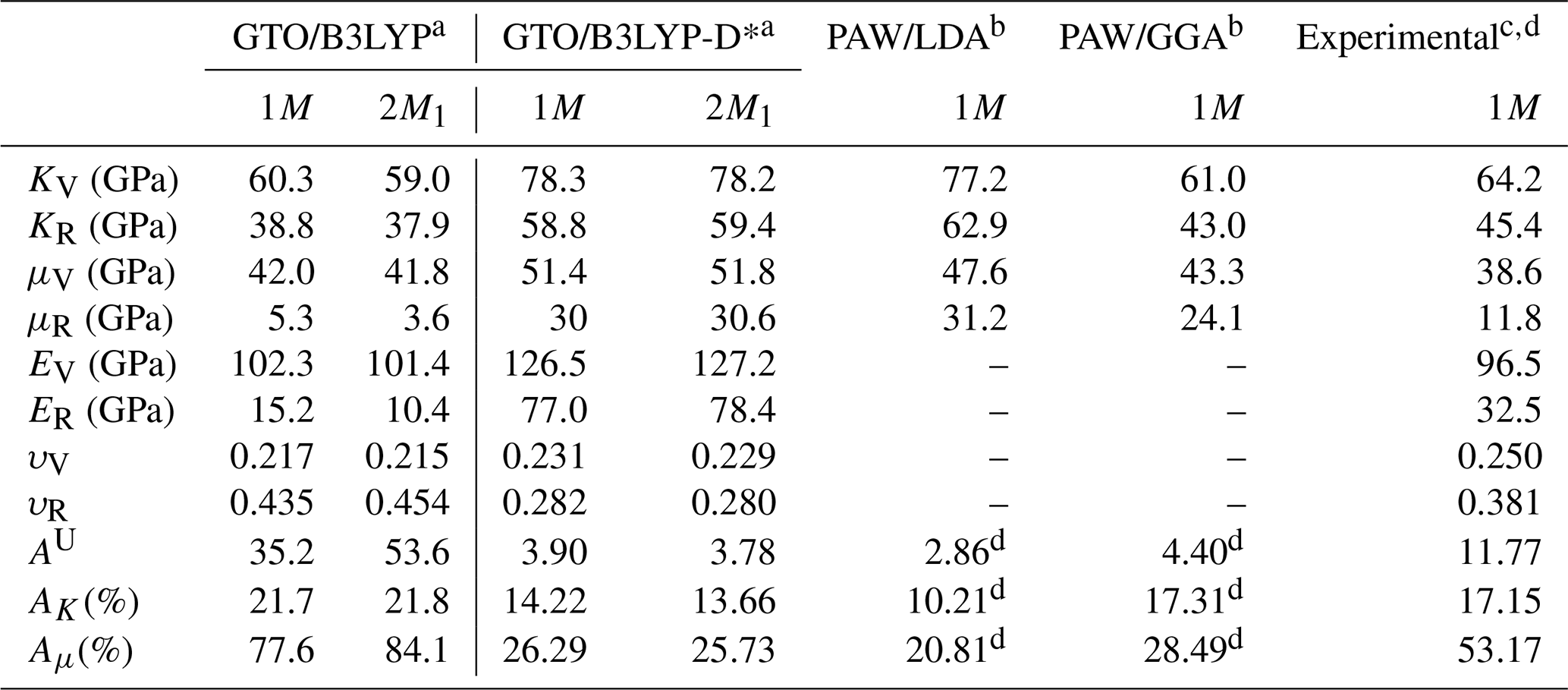
a Present work. b Chheda et al. (2014). c Aleksandrov et al. (1974). Subscripts V and R indicate the Voigt (upper) and Reuss (lower) bounds, respectively. d Values calculated in the present work from the second-order elastic moduli reported by Aleksandrov et al. (1974).
To further assess the validity of the linear model used to calculate the axial compressibilities, the axial moduli M(a), M(b) and M(c) were calculated from the elastic compliance tensor components according to the expressions reported by Mookherjee et al. (2016):
For instance, the calculated moduli were M(a)=344.3, M(b)=365.2 and M(c)=88.0 GPa for phlogopite-1M, and M(a)=360.5, M(b)=353.9 and M(c)=89.1 GPa for the 2M1 polytype, values consistent with those reported in Table 3 obtained from the equation of state fit and that provide another measure of the anisotropy of the mineral, with a ratio for both polytypes.
Directional single-crystal elastic properties (Young's modulus E, linear compressibility β= K−1, shear modulus μ and Poisson's ratio υ) were calculated in the Cartesian space using the QUANTAS code (Gaillac et al., 2016; Ulian and Valdrè, 2022). The interested reader can find in the cited works all the formulations employed to calculate the spatial dependence of the cited properties. Differently from the other elastic properties reported above, where no significant variation between the 1M and 2M1 phlogopite polytypes was evinced, the directional properties showed some effect due to the different TOT stacking (see Fig. S1). For example, the shape of the Young's modulus on the (xy) plane (Fig. S1a) is slightly more compressed along NE-SW and NO-SE directions in the polytype-1M, while it appeared more isotropic in phlogopite-2M1. The shear moduli on the (xz) plane of the latter polytype (see Fig. S1c) is slightly canted, with the oblate maxima direction almost parallel to the east-west direction.
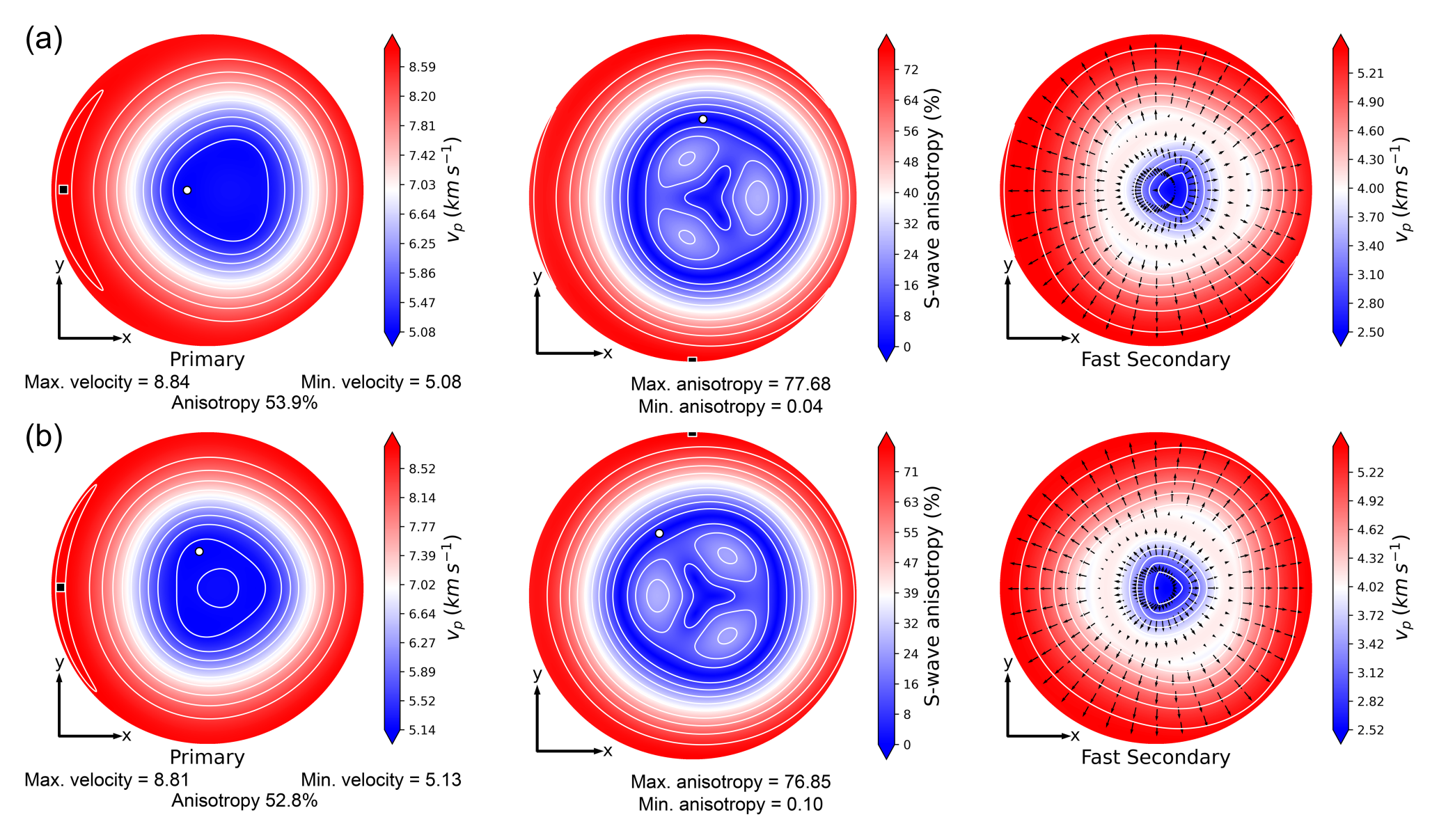
Figure 4Lamber equal area (stereographic) projection of the upper hemisphere of the seismic anisotropy calculated for (a) 1M and (b) 2M1 polytypes of phlogopite. In each panel, the left picture is related to the primary velocity, the central one is the S-wave anisotropy (related to the fast and slow shear waves) and the rightmost stereoplot is the polarization of the vS1 (fast secondary) shear velocity. All the quantities were calculated at the DFT/B3LYP-D* level of theory.
Finally, the phase (seismic) velocities were calculated by solving Christoffel's equation (Musgrave, 1970) using the routines implemented in the QUANTAS code (Jaeken and Cottenier, 2016; Ulian and Valdrè, 2022). The results are shown in Fig. 4, where the compressional wave velocity (vP, P-wave, primary), the anisotropy of the shear velocities (S-wave, secondary), and the polarization of the fast shear wave (vS1) are reported for both phlogopite polytypes. From the stereographic projections, particularly the S-wave anisotropy, it is immediately visible an east-west mirror plane normal to the b-axis that is related to the monoclinic crystal system of the mineral. This figure is in very good agreement with the theoretical analysis of Chheda et al. (2014), with some degree of deviation of the absolute values of the seismic velocities and anisotropy due to the stiffer elastic moduli obtained with the combination of the Gaussian-type orbitals basis sets and hybrid B3LYP-D* functional.
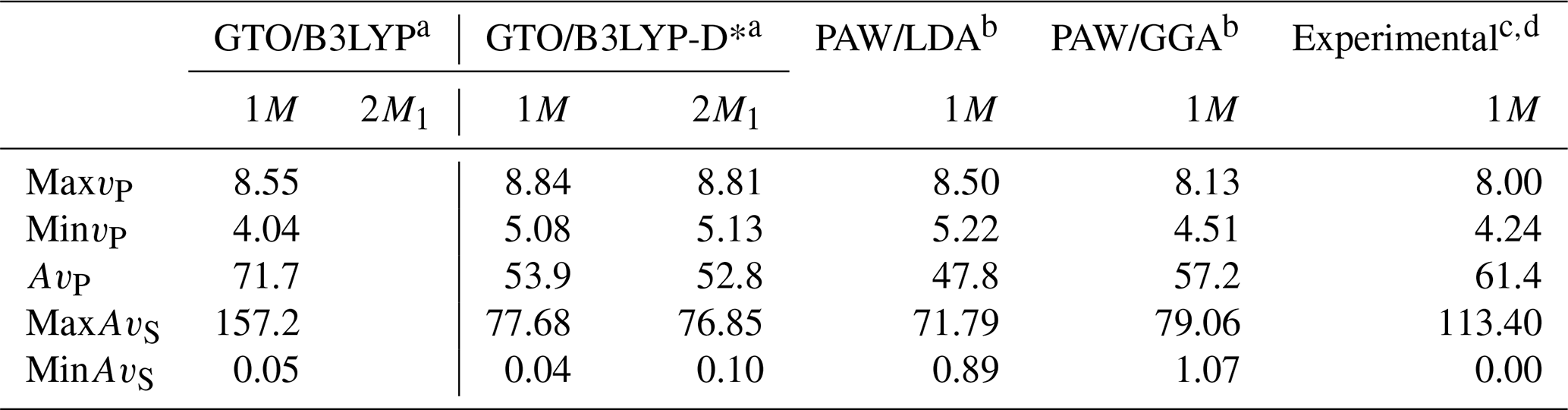
Table 6 reports the maximum and minimum vP values, the percentage of vP anisotropy (AvP) and the range of S-wave anisotropy (AvS), as calculated from the present GTO/B3LYP(-D*) simulations for both phlogopite polytypes, together with previous experimental (Aleksandrov et al., 1974) and theoretical (Chheda et al., 2014) data related to the phlogopite 1M. The results are in general good agreement, with a slight overestimation of the maximum P-wave velocity and a smaller minimum primary wave velocity obtained from the uncorrected B3LYP functional. The shear wave anisotropy at the B3LYP-D* level of theory is within the values obtained by Chheda and collaborators (2014), with the maximum AvS value being underestimated. This latter datum is instead more than double when calculated from the B3LYP results, and very overestimated compared to the experimental value reported by Aleksandrov et al. (1974).
As mentioned in the introduction, the knowledge of the elastic properties could help explain and interpret some geophysical observations reported in previous analyses. For instance, a large delay time (δt) of about 1–2 s was measured in certain subduction zones, like the Ryukyu and Calabrian arcs, in addition to the trench-parallel shear wave splitting (Long and Silver, 2008). Chheda and collaborators (2014) suggested that peridotitic rocks with olivine as the major component would require a very large thickness ranging from 100 to 150 km to justify this large δt value. However, phyllosilicates like talc (Mainprice et al., 2008; Ulian et al., 2014), muscovite (Ulian and Valdrè, 2015a), antigorite (Mookherjee and Capitani, 2011) and phlogopite (Pavese et al., 2003; Chheda et al., 2014) present significantly higher anisotropic behaviours than olivine. Thus, their presence in the subduction layer would require smaller thickness (∼10–15 km) to explain both the trench parallel shear wave polarization anisotropy and the large delay time (Chheda et al., 2014). Indeed, according to previous investigations (Trønnes, 2002), phlogopite has a wide PT stability, thus it can be found in different settings, e.g., in the upper mantle and subduction zone, the latter being also characterised by hydrating conditions that could further stabilise this trioctahedral mica and other hydrous phases.
Table 6Maximum and minimum compressional wave velocity (vP, km s−1), P-wave anisotropy (AvP, %), and maximum and minimum S-wave anisotropy (AvS, %) of phlogopite.
a Present work. b Chheda et al. (2014). c Aleksandrov et al. (1974). d Values calculated in the present work from the second-order elastic moduli reported by Aleksandrov et al. (1974).
In the present work, using ab initio Density Functional Theory simulations, the elastic properties of two phlogopite polytypes, namely the 1M and the 2M1, were investigated considering hydrostatic compression up to about 15 GPa and the monoclinic elastic tensor. The following conclusions can be drawn:
-
despite the different stacking of the TOT layers, the elastic properties of phlogopite were not severely affected by polytypism. Considering the numerical errors of the fitting procedure, the bulk modulus K0 obtained from the third-order Birch-Murnaghan equation of state was in the range 55–60 GPa for both polytypes.
-
the compressional behaviour of phlogopite is highly anisotropic, with the a and b crystallographic axes being about four times less compressible than the c one, as noticed from the linear moduli. This agrees with the experimental observations made on dioctahedral and trioctahedral micas, and it is due to several factors, i.e., the high compressibility of the interlayer region, the low compressibility of the tetrahedral and octahedral polyhedral, and the accommodation of the stress within the TOT layers via variation of the tetrahedral rotation angle αr.
-
the elastic tensors of phlogopite-1M and phlogopite-2M1 have comparable Cij components (in Voigt's notation), with a difference in sign and absolute value in the C25 one. This is probably the reason of the small differences observed in the directionally dependent elastic properties.
-
the DFT simulations were able to correctly describe the behaviour of the known 1M polytype of phlogopite, extending the knowledge to the 2M1 one. In particular, the use of the DFT-D2 correction provided a robust description of the physical forces acting along [001], namely the stacking direction of the TOT +i layers.
It is also known that the elastic properties of micas can be deeply affected by the presence of cationic/anionic substitutions in the T and O sheets, e.g., FeII substituting MgII. These are currently under investigation and will be the subject of a future work.
From the mineralogical, petrological, and geophysical perspectives, the knowledge of the physical properties of phlogopite is of utmost importance for deepening the understanding of the thermodynamic and kinetic stability of altered rock assemblages, such as the mica–amphibole–rutile–ilmenite–diopside system. Indeed, the phlogopite chemical composition is similar to that of the ternary system olivine–quartz–alkali, which is also close to the bulk composition of the cited altered rocks. Also, the elasticity of the trioctahedral mica is relevant both for bulk and 2D applications of phlogopite in materials science, owing to the dielectric properties of the mineral that can be tuned by the exfoliation in the basal plane and cationic substitutions, particularly in the octahedral sheet. First principles simulations can help in shedding light on these topics, providing useful resources for the interpretation of geophysical phenomena and the development of more, sustainable materials for advanced applications.
The results of the present work are reported in the manuscript and in a dedicated dataset published at the following link: https://doi.org/10.17632/r4wmxz7kcc.1 (Ulian and Valdrè, 2024c).
The supplement related to this article is available online at https://doi.org/10.5194/se-17-601-2026-supplement.
Conceptualization, G.U. and G.V.; methodology, G.U. and F.R.; validation, G.U. and G.V.; formal analysis, G.U.; investigation, G.U. and G.V.; data curation, G.U. and F.R.; writing–review and editing, G.U., F.R. and G.V.; visualization, G.U. and F.R.; supervision, G.V. All authors have read and agreed to the published version of the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
The authors wish to thank the University of Bologna for supporting the present research. All the simulations were performed with the computational resources (HPC cluster) and software license of the Interdisciplinary Research Centre of Biomineralogy, Crystallography and Biomaterials, Department of Biological, Geological and Environmental Sciences, University of Bologna.
This paper was edited by Andrea Di Muro and reviewed by Yunguo Li and one anonymous referee.
Alexandrov, K. S. and Ryzhova, T. V.: Elastic properties of rock-forming minerals. II. Layered silicates, Bulletin of the Academy of Sciences of the U.S.S.R, Geophysics Series, 9, 1165–1168, 1961.
Aleksandrov, K. S., Alchikov, U. V., Belikov, B. P., Zaslavskii, B. I., and Krupnyi, A. I.: Velocities of elastic waves in minerals at atmospheric pressure and increasing precision of elastic constants by means of EVM, in: Izv. Acad. Sci. USSR, Geol. Ser., 10, 15–24, 1974 (in Russian).
Angel, R. J., Gonzalez-Platas, J., and Alvaro, M.: EosFit7c and a Fortran module (library) for equation of state calculations, Zeitschrift Fur Kristallographie, 229, 405–419, https://doi.org/10.1515/zkri-2013-1711, 2014.
Ariane, K., Tamayo, A., Chorfa, A., Rubio, F., and Rubio, J.: Optimization of the nucleating agent content for the obtaining of transparent fluormica glass-ceramics, Ceramics International, 49, 9826–9838, https://doi.org/10.1016/j.ceramint.2022.11.156, 2023.
Becke, A. D.: Density-Functional Thermochemistry .3. The Role of Exact Exchange, Journal of Chemical Physics, 98, 5648–5652, https://doi.org/10.1063/1.464913, 1993.
Belmonte, D.: First Principles Thermodynamics of Minerals at HP-HT Conditions: MgO as a Prototypical Material, Minerals, 7, 183, https://doi.org/10.3390/Min7100183, 2017.
Birch, F.: Finite elastic strain of cubic crystal, Phys. Rev., 71, 809–824, 1947.
Brandenburg, J. G., Alessio, M., Civalleri, B., Peintinger, M. F., Bredow, T., and Grimme, S.: Geometrical correction for the inter- and intramolecular basis set superposition error in periodic density functional theory calculations, Journal of Physical Chemistry A, 117, 9282–9292, https://doi.org/10.1021/jp406658y, 2013.
Broyden, C. G.: The convergence of a class of double-rank minimization algorithms: 1. General considerations, IMA Journal of Applied Mathematics, 6, 76–90, https://doi.org/10.1093/imamat/6.1.76, 1970a.
Broyden, C. G.: The convergence of a class of double-rank minimization algorithms: 2. The new algorithm, IMA Journal of Applied Mathematics, 6, 222–231, https://doi.org/10.1093/imamat/6.3.222, 1970b.
Cadore, A. R., De Oliveira, R., Longuinhos, R., Teixeira, V. D. C., Nagaoka, D. A., Alvarenga, V. T., Ribeiro-Soares, J., Watanabe, K., Taniguchi, T., Paniago, R. M., Malachias, A., Krambrock, K., Barcelos, I. D., and De Matos, C. J. S.: Exploring the structural and optoelectronic properties of natural insulating phlogopite in van der Waals heterostructures, 2D Materials, 9, 035007, https://doi.org/10.1088/2053-1583/ac6cf4, 2022.
Catti, M., Ferraris, G., Hull, S., and Pavese, A.: Powder Neutron-Diffraction Study of 2M1 Muscovite at Room Pressure and at 2 Gpa, European Journal of Mineralogy, 6, 171–178, 1994.
Chheda, T. D., Mookherjee, M., Mainprice, D., dos Santos, A. M., Molaison, J. J., Chantel, J., Manthilake, G., and Bassett, W. A.: Structure and elasticity of phlogopite under compression: Geophysical implications, Physics of the Earth and Planetary Interiors, 233, 1–12, https://doi.org/10.1016/j.pepi.2014.05.004, 2014.
Civalleri, B., Zicovich-Wilson, C. M., Valenzano, L., and Ugliengo, P.: B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals, CrystEngComm, 10, 405–410, https://doi.org/10.1039/b715018k, 2008.
Comodi, P. and Zanazzi, P. F.: High-Pressure Structural Study of Muscovite, Physics and Chemistry of Minerals, 22, 170–177, 1995.
Comodi, P., Fumagalli, P., Montagnoli, M., and Zanazzi, P. F.: A single-crystal study on the pressure behavior of phlogopite and petrological implications, American Mineralogist, 89, 647–653, https://doi.org/10.2138/am-2004-0420, 2004.
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C., and Lundqvist, B. I.: Van der Waals Density Functional for General Geometries, Physical Review Letters, 92, 246401, https://doi.org/10.1103/PhysRevLett.92.246401, 2004.
Donnay, G., Morimoto, N., and Takeda, H.: Trioctahedral one-layer micas. II. Prediction of the structure from composition and cell dimensions, Acta Cryst., 17, 1374–1381, https://doi.org/10.1107/S0365110X64003462, 1964a.
Donnay, G., Morimoto, N., and Takeda, H.: Trioctahedral one-layer micas. I. Crystal structure of a synthetic iron mica, Acta Cryst., 17, 1369–1373, https://doi.org/10.1107/S0365110X64003450, 1964b.
Dovesi, R., Roetti, C., Freyria Fava, C., Prencipe, M., and Saunders, V. R.: On the elastic properties of lithium, sodium an potassium oxide. An ab initio study, Chemical Physics, 156, 11–19, 1991.
Dovesi, R., Erba, A., Orlando, R., Zicovich-Wilson, C. M., Civalleri, B., Maschio, L., Rerat, M., Casassa, S., Baima, J., Salustro, S., and Kirtman, B.: Quantum-mechanical condensed matter simulations with CRYSTAL, Wiley Interdisciplinary Reviews-Computational Molecular Science, 8, E1360, https://doi.org/10.1002/Wcms.1360, 2018.
Fletcher, R.: A new approach to variable metric algorithms, The Computer Journal, 13, 317–322, https://doi.org/10.1093/comjnl/13.3.317, 1970.
Gaillac, R., Pullumbi, P., and Coudert, F. X.: ELATE: an open-source online application for analysis and visualization of elastic tensors, Journal of Physics-Condensed Matter, 28, 275201, https://doi.org/10.1088/0953-8984/28/27/275201, 2016.
Gatta, G. D., Merlini, M., Rotiroti, N., Curetti, N., and Pavese, A.: On the crystal chemistry and elastic behavior of a phlogopite 3T, Physics and Chemistry of Minerals, 38, 655–664, https://doi.org/10.1007/s00269-011-0438-z, 2011.
Gatta, G. D., Lotti, P., Merlini, M., Liermann, H.-P., Lausi, A., Valdrè, G., and Pavese, A.: Elastic behaviour and phase stability of pyrophyllite and talc at high pressure and temperature, Physics and Chemistry of Minerals, 42, 309–318, https://doi.org/10.1007/s00269-014-0721-x, 2015.
Gatti, C., Saunders, V. R., and Roetti, C.: Crystal-field effects on the topological properties of the electron-density in molecular-crystals – the case of urea, Journal of Chemical Physics, 101, 10686–10696, https://doi.org/10.1063/1.467882, 1994.
Geng, X., Tian, S., Xu, W., Chen, L., Lu, N., Liang, Z., Hu, W., and Liu, J.: A Two-Stage Geodynamic Model for Post-Collisional Potassic-Ultrapotassic Magmatism in Southeast Tibet, Journal of Geophysical Research: Solid Earth, 129, https://doi.org/10.1029/2024JB028887, 2024.
Goldfarb, D.: A family of variable-metric methods derived by variational means, Mathematics of Computation, 24, 23–26, https://doi.org/10.1090/S0025-5718-1970-0258249-6 1970.
Grimme, S.: Semiempirical GGA-type density functional constructed with a long-range dispersion correction, Journal of Computational Chemistry, 27, 1787–1799, https://doi.org/10.1002/jcc.20495, 2006.
Guidotti, C. V. and Sassi, F. P.: Muscovite as a petrogenetic indicator mineral in pelitic schists, Neues Jahrbuch fuer Mineralogie, Abhandlungen, 123, 97–142, 1976.
Hebbache, M. and Zemzemi, M.: Ab initio study of high-pressure behavior of a low compressibility metal and a hard material: Osmium and diamond, Physical Review B, 70, https://doi.org/10.1103/Physrevb.70.224107, 2004.
Hill, R.: The elastic behaviour of a crystalline aggregate, Proceedings of the Physical Society, London, Section A, 65, 349–354, 1952.
Icenhower, J. and London, D.: An experimental study of element partitioning among biotite, muscovite, and coexisting peraluminous silicic melt at 200 MPa (H2O), American Mineralogist, 80, 1229–1251, https://doi.org/10.2138/am-1995-11-1213, 1995.
Jaeken, J. W. and Cottenier, S.: Solving the Christoffel equation: Phase and group velocities, Computer Physics Communications, 207, 445–451, https://doi.org/10.1016/j.cpc.2016.06.014, 2016.
King, T. T., Grayeski, W., and Cooper, R. F.: Thermochemical reactions and equilibria between fluoromicas and silicate matrices: A petromimetic perspective on structural ceramic composites, Journal of the American Ceramic Society, 83, 2287–2296, https://doi.org/10.1111/j.1151-2916.2000.tb01549.x, 2000.
Kruse, H. and Grimme, S.: A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems, Journal of Chemical Physics, 136, https://doi.org/10.1063/1.3700154, 2012.
Lacalamita, M., Mesto, E., Scordari, F., and Schingaro, E.: Chemical and structural study of 1M- and 2M (1)-phlogopites coexisting in the same Kasenyi kamafugitic rock (SW Uganda), Physics and Chemistry of Minerals, 39, 601–611, https://doi.org/10.1007/s00269-012-0515-y, 2012.
Laurora, A., Brigatti, M. F., Mottana, A., Malferrari, D., and Caprilli, E.: Crystal chemistry of trioctahedral micas alkaline and subalkaline volcanic rocks: A case study from Mt. Sassetto (Tolfa district, central Italy), American Mineralogist, 92, 468–480, https://doi.org/10.2138/am.2007.2339, 2007.
Lee, C. T., Yang, W. T., and Parr, R. G.: Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density, Physical Review B, 37, 785–789, https://doi.org/10.1103/PhysRevB.37.785, 1988.
Long, M. D. and Silver, P. G.: The subduction zone flow field from seismic anisotropy: a global view, Science, 319, 315–318, https://doi.org/10.1126/science.1150809, 2008.
Mainprice, D., Le Page, Y., Rodgers, J., and Jouanna, P.: Ab initio elastic properties of talc from 0 to 12 GPa: Interpretation of seismic velocities at mantle pressures and prediction of auxetic behaviour at low pressure, Earth and Planetary Science Letters, 274, 327–338, https://doi.org/10.1016/j.epsl.2008.07.047, 2008.
Melzer, S. and Wunder, B.: K-Rb-Cs partitioning between phlogopite and fluid: Experiments and consequences for the LILE signatures of island arc basalts, Lithos, 59, 69–90, https://doi.org/10.1016/S0024-4937(01)00061-5, 2001.
Momma, K. and Izumi, F.: VESTA: a three-dimensional visualization system for electronic and structural analysis, J. Appl. Crystallogr., 41, 653–658, 2008.
Mookherjee, M. and Capitani, G. C.: Trench parallel anisotropy and large delay times: Elasticity and anisotropy of antigorite at high pressures, Geophys. Res. Lett., 38, L09315, https://doi.org/10.1029/2011gl047160, 2011.
Mookherjee, M., Tsuchiya, J., and Hariharan, A.: Crystal structure, equation of state, and elasticity of hydrous aluminosilicate phase, topaz-OH (Al2SiO4(OH)(2)) at high pressures, Physics of the Earth and Planetary Interiors, 251, 24–35, https://doi.org/10.1016/j.pepi.2015.11.006, 2016.
Musgrave, M. J. P.: Crystal Acoustics: introduction to the study of elastic waves and vibrations in crystals, Holden-Day, San Francisco, CA, USA, ISBN 9780816262021, 1970.
Nada, R., Nicholas, J. B., McCarthy, M. I., and Hess, A. C.: Basis sets for ab initio periodic Hartree-Fock studies of zeolite/adsorbate interactions: He, Ne, and Ar in silica sodalite, International Journal of Quantum Chemistry, 60, 809-820, https://doi.org/10.1002/(SICI)1097-461X(1996)60:4<809::AID-QUA3>3.0.CO;2-0, 1996.
Nye, J. F.: Physical properties of crystals, Oxford University Press, Oxford, ISBN 9780198511656., 1957.
Ottonello, G., Civalleri, B., Ganguly, J., Perger, W. F., Belmonte, D., and Zuccolini, M. V.: Thermo-chemical and thermo-physical properties of the high-pressure phase anhydrous B (Mg14Si5O24): An ab-initio all-electron investigation, American Mineralogist, 95, 563–573, https://doi.org/10.2138/am.2010.3368, 2010.
Pavese, A., Ferraris, G., Pischedda, V., and Mezouar, M.: Synchrotron powder diffraction study of phengite 3T from the Dora-Maira massif: P-V-T equation of state and petrological consequences, Physics and Chemistry of Minerals, 26, 460–467, https://doi.org/10.1007/s002690050208, 1999.
Pavese, A., Levy, D., Curetti, N., Diella, V., Fumagalli, P., and Sani, A.: Equation of state and compressibility of phlogopite by in-situ high-pressure X-ray powder diffraction, European Journal of Mineralogy, 15, 455–463, https://doi.org/10.1127/0935-1221/2003/0015-0455, 2003.
Pawley, A. R., Clark, S. M., and Chinnery, N. J.: Equation of state measurements of chlorite, pyrophyllite, and talc, American Mineralogist, 87, 1172–1182, 2002.
Perger, W. F., Criswell, J., Civalleri, B., and Dovesi, R.: Ab-initio calculation of elastic constants of crystalline systems with the CRYSTAL code, Computer Physics Communications, 180, 1753–1759, https://doi.org/10.1016/j.cpc.2009.04.022, 2009.
Pfrommer, B. G., Côté, M., Louie, S. G., and Cohen, M. L.: Relaxation of Crystals with the Quasi-Newton Method, Journal of Computational Physics, 131, 233–240, https://doi.org/10.1006/jcph.1996.5612, 1997.
Prencipe, M., Noel, Y., Bruno, M., and Dovesi, R.: The vibrational spectrum of lizardite-1T [Mg(3)Si(2)O(5)(OH)(4)] at the Gamma point: A contribution from an ab initio periodic B3LYP calculation, American Mineralogist, 94, 986–994, 2009.
Ranganathan, S. I. and Ostoja-Starzewski, M.: Universal elastic anisotropy index, Physical Review Letters, 101, 055504, https://doi.org/10.1103/PhysRevLett.101.055504, 2008.
Shanno, D. F.: Conditioning of quasi-Newton methods for function minimization, Mathematics of Computation, 24, 647–656, https://doi.org/10.1090/S0025-5718-1970-0274029-X 1970.
Sudo, A. and Tatsumi, Y.: Phlogopite and K-amphibole in the upper mantle: Implication for magma genesis in subduction zones, Geophys. Res. Lett., 17, 29–32, https://doi.org/10.1029/GL017i001p00029, 1990.
Sweeney, R. J., Thompson, A. B., and Ulmer, P.: Phase relations of a natural MARID composition and implications for MARID genesis, lithospheric melting and mantle metasomatism, Contributions to Mineralogy and Petrology, 115, 225–241, https://doi.org/10.1007/BF00321222, 1993.
Trønnes, R. G.: Stability range and decomposition of potassic richterite and phlogopite end members at 5-15 GPa, Mineralogy and Petrology, 74, 129–148, https://doi.org/10.1007/s007100200001, 2002.
Tutti, F., Dubrovinsky, L. S., and Nygren, M.: High-temperature study and thermal expansion of phlogopite, Physics and Chemistry of Minerals, 27, 599–603, https://doi.org/10.1007/s002690000098, 2000.
Ulian, G. and Valdrè, G.: Density functional investigation of the thermo-physical and thermo-chemical properties of 2M(1) muscovite, American Mineralogist, 100, 935–944, https://doi.org/10.2138/am-2015-5086, 2015a.
Ulian, G. and Valdrè, G.: Structural, vibrational and thermophysical properties of pyrophyllite by semi-empirical density functional modelling, Physics and Chemistry of Minerals, 42, 609–627, https://doi.org/10.1007/s00269-015-0748-7, 2015b.
Ulian, G. and Valdrè, G.: Density functional investigation of the thermophysical and thermochemical properties of talc Mg3Si4O10(OH)(2), Physics and Chemistry of Minerals, 42, 151–162, https://doi.org/10.1007/s00269-014-0708-7, 2015c.
Ulian, G. and Valdrè, G.: Second-order elastic constants of hexagonal hydroxylapatite (P63) from ab initio quantum mechanics: comparison between DFT functionals and basis sets, International Journal of Quantum Chemistry, 118, e25500, https://doi.org/10.1002/qua.25500, 2018.
Ulian, G. and Valdrè, G.: Equation of state and second-order elastic constants of portlandite Ca(OH)2 and brucite Mg(OH)2, Physics and Chemistry of Minerals, 46, 101–117, https://doi.org/10.1007/s00269-018-0989-3, 2019.
Ulian, G. and Valdrè, G.: QUANTAS, a Python software for the analysis of solids from ab initio quantum mechanical simulations and experimental data, J. Appl. Crystallogr., 55, 386–396, https://doi.org/10.1107/S1600576722000085, 2022.
Ulian, G. and Valdrè, G.: Crystal-chemical, vibrational and electronic properties of 1M-phlogopite K(Mg,Fe)3Si3AlO10(OH)2 from Density Functional Theory simulations, Applied Clay Science, 246, 107166, https://doi.org/10.1016/j.clay.2023.107166, 2023a.
Ulian, G. and Valdrè, G.: The effect of long-range interactions on the infrared and Raman spectra of aragonite (CaCO3, Pmcn) up to 25 GPa, Scientific Reports, 13, 2725, https://doi.org/10.1038/s41598-023-29783-7, 2023b.
Ulian, G. and Valdrè, G.: SEISMIC, a Python-based code of the QUANTAS package to calculate the phase and group acoustic velocities in crystals, Computers and Geosciences, 188, https://doi.org/10.1016/j.cageo.2024.105615, 2024a.
Ulian, G. and Valdrè, G.: Crystal structure and elastic and phonon properties of realgar versus pressure, J. Appl. Crystallogr., 57, 220–231, https://doi.org/10.1107/S1600576724000025, 2024b.
Ulian, G. and Valdrè, G.: Elastic properties of phlogopite 1M and 2M1 polytypes, Mendeley Data, V1 [data set], https://doi.org/10.17632/r4wmxz7kcc.1, 2024c.
Ulian, G. and Valdrè, G.: Crystallographic, electronic and vibrational properties of 2D silicate monolayers, J. Appl. Crystallogr., 58, 349–362, https://doi.org/10.1107/S1600576725000731, 2025.
Ulian, G., Tosoni, S., and Valdrè, G.: Comparison between Gaussian-type orbitals and plane wave ab initio density functional theory modeling of layer silicates: Talc Mg3Si4O10(OH)(2) as model system, Journal of Chemical Physics, 139, 204101, https://doi.org/10.1063/1.4830405, 2013.
Ulian, G., Tosoni, S., and Valdrè, G.: The compressional behaviour and the mechanical properties of talc [Mg3Si4O10(OH)2]: a density functional theory investigation, Physics and Chemistry of Minerals, 41, 639–650, https://doi.org/10.1007/s00269-014-0677-x, 2014.
Ulian, G., Moro, D., and Valdrè, G.: Elastic properties of heterodesmic composite structures: The case of calcite CaCO3 (space group R-3c), Composites Part C: Open Access, 6, https://doi.org/10.1016/j.jcomc.2021.100184, 2021.
Valenzano, L., Torres, F. J., Doll, K., Pascale, F., Zicovich-Wilson, C. M., and Dovesi, R.: Ab initio study of the vibrational spectrum and related properties of crystalline compounds; the case of CaCO3 calcite, Zeitschrift Fur Physikalische Chemie-International Journal of Research in Physical Chemistry & Chemical Physics, 220, 893–912, https://doi.org/10.1524/zpch.2006.220.7.893, 2006.
Valenzano, L., Noel, Y., Orlando, R., Zicovich-Wilson, C. M., Ferrero, M., and Dovesi, R.: Ab initio vibrational spectra and dielectric properties of carbonates: magnesite, calcite and dolomite, Theoretical Chemistry Accounts, 117, 991–1000, https://doi.org/10.1007/s00214-006-0213-2, 2007.
Van Reenen, D. D., Smit, C. A., Huizenga, J. M., Tsunogae, T., and Safonov, O.: Thermo-tectonic evolution of the Neoarchaean Southern Marginal Zone of the Limpopo granulite Complex (South Africa), South African Journal of Geology, 126, 373–406, https://doi.org/10.25131/sajg.126.0027, 2023.
Vaughan, M. T. and Guggenheim, S.: Elasticity of muscovite and its relationship to crystal structure, Journal of Geophysical Research: Solid Earth, 91, 4657–4664, https://doi.org/10.1029/JB091iB05p04657, 1986.
Ventruti, G., Levy, D., Pavese, A., Scordari, F., and Suard, E.: High-temperature treatment, hydrogen behaviour and cation partitioning of a Fe-Ti bearing volcanic phlogopite by in situ neutron powder diffraction and FTIR spectroscopy, European Journal of Mineralogy, 21, 385–396, https://doi.org/10.1127/0935-1221/2009/0021-1903, 2009.
Virgo, D. and Popp, R. K.: Hydrogen deficiency in mantle-derived phlogopites, American Mineralogist, 85, 753–759, https://doi.org/10.2138/am-2000-5-614, 2000.
Wang, J., Wang, Q., Ma, L., Hu, W. L., Wang, J., Belousova, E., and Tang, G. J.: Rapid Recycling of Subducted Sediments in the Subcontinental Lithospheric Mantle, Journal of Petrology, 64, https://doi.org/10.1093/petrology/egad056, 2023.